Cellular Mechanism, Ubiquitination/proteasome
Product Name: LDN-57444 | UCH-L1inhibitor (#C5574-5)
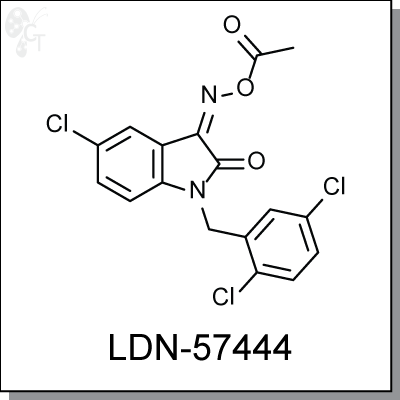
LDN-57444 is a specific inhibitor (Ki = 0.4 ?M) against Ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), a
member of deubiquitinating enzymes (DUBs). LDN-57444 increased proliferation of SH-SY5Y cells, a UCH-
L1-expressing neuroblastoma line. [1]
As expression of UCH-L1 is highly specific to neurons and to cells of the diffuse neuroendocrine system and
their tumors, LDN-57444 is a useful tool for studying the roles of UCH-L1 in Alzheimers disease, Parkinsons
disease, and other neurological disorders. [2,3]
|
Details
|
Chemical Formula:
|
|
C17H11CI3N2O3
|
|
CAS No.:
|
|
668467-91-2
|
|
Molecular weight:
|
|
397.64
|
|
Purity:
|
|
> 98%
|
|
Appearance:
|
|
Yellow
|
|
Chemical name:
|
|
1H-Indole-2,3-dione, 5-chloro-1-[(2,5-dichlorophenyl)methyl]-, 3-(O-acetyloxime)
|
|
Solubility:
|
|
Up to 25 mM in DMSO
|
|
Synonyms:
|
|
LDN-57444, LDN57444
|
|
Storage:
|
|
For longer shelf life, store solid powder at 4oC desiccated, or store DMSO solution
at -20oC
|
1. Liu Y, et al. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell
line. Chem Biol. 2003; 10(9):837-46. Pubmed ID: 14522054
2. Cartier AE, et al. Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J Neurosci. 2009; 29
(24):7857-68. Pubmed ID: 19535597
3. Zhang M, et al. Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal
hydrolase L1. J Neurochem. 2012; 120(6):1129-38. Pubmed ID: 22212137
|
Product Name: MG132 | Proteasome inhibitor (#C6413-5)
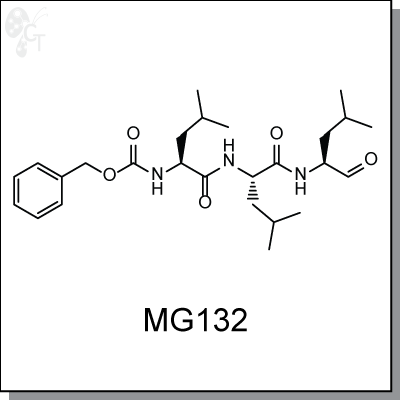
MG-132 is a triterpene, peptide-aldehyde proteasome inhibitor derived from a Chinese medicinal plant. It
inhibits 20S proteasome activty by covalently binding to the active site of the beta subunits and effectively
blocks the proteolytic activity of the 26S proteasome complex. Through the formation of reactive oxygen
species, MG-132 inhibits tumor cell growth by inducing cell cycle arrest and triggering apoptosis. [1]
MG-132 was found to enhance IL-6 expression in HUVEC. MG-132 does not affect IkB levels, suggesting that
proteasome inhibition does not influence the NF-kB system without any agonist stimulation. In similar
studies, MG-132 is believed to inhibit the degradation of phosphorylated MEK1/2 and activate the
downstream region of this signalling cascade. [2]
In NCIH2452 and NCI H2052 human thoracic malignant pleural mesothelioma (MPM) cell lines, it was foud
that 0.5 uM concentrations of MG-132 caused significant apoptosis as evidenced by DNA damage, cleavage
of PARP and caspases 3, 7, and 9. [3]
|
Details
|
Chemical Formula:
|
|
C26H41N3O5
|
|
CAS No.:
|
|
133407-82-6
|
|
Molecular weight:
|
|
475.62
|
|
Purity:
|
|
> 98%
|
|
Appearance:
|
|
White
|
|
Chemical name:
|
|
benzyl (S)-4-methyl-1-((S)-4-methyl-1-((S)-4-methyl-1-oxopentan-2-ylamino)-1-
oxopentan-2-ylamino)-1-oxopentan-2-ylcarbamate
|
|
Solubility:
|
|
Up to 100 mM in DMSO
|
|
Synonyms:
|
|
MG-132, MG132, Z-LLL-CHO, MG 132
|
|
Storage:
|
|
For longer shelf life, store solid powder at 4oC desiccated, or store DMSO solution
at -20oC
|
References
1. Guo et al., MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac. J. Oncol. 2012,
doi:10.1111/j.1743-7563.2012.01535.x
2. Shibata et al., Proteasome inhibitor MG-132 enhances the expression of interleukin-6 in human umbilical
vein endothelial cells: Involvement of MAP/ERK kinase. Immunol. Cell Biol. 2002, 80, 226-230.
Pubmed ID: 12067409
3. Yuan et al., Proteasome Inhibitor MG132 Induces Apoptosis and Inhibits Invasion of Human Malignant
Pleural Mesothelioma Cells. Translational Oncol. 2008, 1(3), 129-140. Pubmed ID: 18795123
|
Product Name: P22077 | USP7 inhibitor (#C7220)
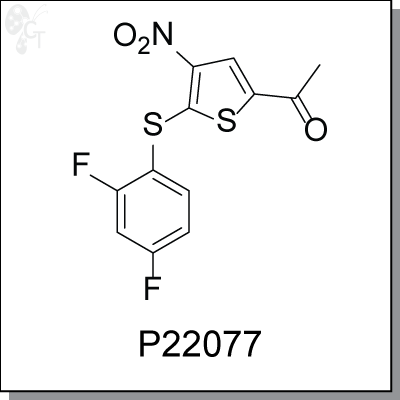
P22077 is a potent and selective USP7 inhibitor. It specifically inhibits deubiquitylating enzyme (DUB) USP7
and the closely related USP47 (at 5µM), but not the other majority of DUBs (at 15-45µM). USP7 has been
found to be critical in cancer progression due to its influence on the stability of the tumor suppressor p53.
The inhibition of USP7 is predicted to destabilize HDM2 and stabilize p53, and USP7 exerts both p53-
dependent and p53-independent effects on controlling cell proliferation and apoptosis.
|
Details
|
Chemical Formula:
|
|
C12H7F2NO3S2
|
|
CAS No.:
|
|
1247819-59-5
|
|
Molecular weight:
|
|
315.32
|
|
Purity:
|
|
> 98%
|
|
Appearance:
|
|
White
|
|
Chemical name:
|
|
1-(5-((2,4-difluorophenyl)thio)-4-nitrothiophen-2-yl)ethanone
|
|
Solubility:
|
|
Up to 50 mM in DMSO
|
|
Synonyms:
|
|
TMP195
|
|
Storage:
|
|
For longer shelf life, store solid powder at 4oC desiccated, or store DMSO solution
at -20oC
|
References
1. Mikael Altun et al. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for
Deubiquitylating Enzymes. (2011) Chemistry & Biology 18, 14011412.
|
Product Name: PS-341 (Bortezomib) | Proteasome inhibitor (#C7734-5)
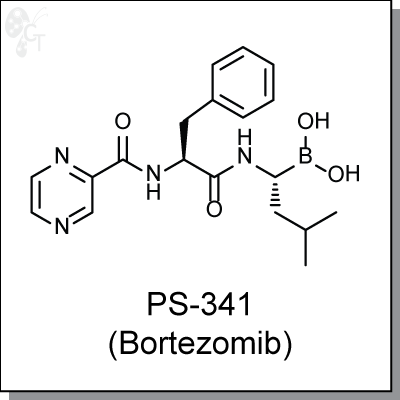
Bortezomib is a first-in-class dipeptide boronic acid-based, water-soluble proteasome inhibitor. As a single
agent, bortezomib was found to have consistent antitumor activity in both chemosensitive and
chemoresistant multiple myeloma cells at an IC50 of 10-20 ng/mL. [1] Bortezomib overcomes the resistance
to apoptosis in multiple myeloma cells that is induced by IL-6. [2] Additionally, bortezomib prevents TNF-a-
induced, NF-kB-dependent upregulation of IL-6 and reduces cell adhesion; proliferation of remaining
adherent multiple myeloma cells was also inhibited by bortezomib. [1]
In MM cell lines U266, IM-9, and Hs Sultan, bortezomib inhibited at IC50 concentrations of 3, 6, and 20 nM,
respectively. [2] Cell growth of Dox40, MR20, and LR5 MM cells was completely inhibited by bortezomib at
100 nM IC50.
Bortezomib suppresses growth and induces apoptosis in Bcr/Abl-positive cells sensitive and resistant to IM.
nterestingly, sequential combination of bortezomib followed by imatinib resulted in a synergistic pro-
apoptotic effect in imatinib-resistant cells; simultaneous exposure of bortezomib and imatinib was
antagonistic. [3]
|
Details
|
Chemical Formula:
|
|
C19H25BN4O4
|
|
CAS No.:
|
|
179324-69-7
|
|
Molecular weight:
|
|
384.24
|
|
Purity:
|
|
> 98%
|
|
Appearance:
|
|
Light Yellow Crystalline
|
|
Chemical name:
|
|
[(1R)-3-methyl-1-({(2S)-3-phenyl-2-[(pyrazin-2-ylcarbonyl)amino]propanoyl}
amino)butyl]boronic acid
|
|
Solubility:
|
|
Up to 100 mM in DMSO
|
|
Synonyms:
|
|
PS-341, PS341, Bortezomib, Velcade, LDP-341
|
|
Storage:
|
|
For longer shelf life, store solid powder at 4oC desiccated, or store DMSO solution
at -20oC
|
References
1. Richardson et al., Cancer Control, 2003, 10(6), 361-369.
2. Hideshima et al., The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes
drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071-3076. Pubmed ID: 1306489
3. Gatto et al., The proteasome inhibitor PS-341 inhibits growth and induces apoptosis in Bcr/Abl-positive
cell lines sensitive and resistant to imatinib mesylate. Haematologica, 2003, 88(8), 853-863.
Pubmed ID: 12935973
|
Product Name: SU5416 (Semaxinib) | Flk-1/KDR inhibitor (#C7541-5)
.png)
SU5416 (Semaxinib) is a reversible, ATP-competitive, oxindole-based inhibitor of Flk-1/KDR receptor tyrosine
kinase. SU5416 inhibits VEGF-dependent phosphorylation of the Flk-1 receptor in Flk-1-overexpressing NIH
3T3 cells with a IC50 of 1.04 uM. In an ELISA-based assay, SU5416 inhibits autophosphorylation of the Flk-1
receptor at an IC50 of 1.23 uM. [1]
In a mitogenic/proliferation assay in HUVECs, the IC50 of SU5416 is extremely time-dependent, ranging from
1 uM to 40 nM at 48h incubation. These data in conjunction with dose-ranging studies support regimens of
less than once-daily dosing of SU5416. [2]
More recent studies have shown that SU5416 is also an agonist of the aryl hydrocarbon receptor (AHR),
leading to the generation of regulatory T-cells in vitro. SU5416 also upregulates CYP1A1 and CYP1B1. [3]
|
Details
|
Chemical Formula:
|
|
C15H14N2O
|
|
CAS No.:
|
|
204005-46-9
|
|
Molecular weight:
|
|
238.28
|
|
Purity:
|
|
> 98%
|
|
Appearance:
|
|
White
|
|
Chemical name:
|
|
2H-Indol-2-one, 3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylene]-1,3-dihydro-, (3Z)
|
|
Solubility:
|
|
Up to 20 mM in DMSO
|
|
Synonyms:
|
|
SU5416, SU 5416, su-5416, Semaxinib
|
|
Storage:
|
|
For longer shelf life, store solid powder at 4oC desiccated, or store DMSO solution
at -20oC
|
References
1. Fong et al., SU5416 Is a Potent and Selective Inhibitor of the Vascular Endothelial Growth Factor Receptor
(Flk-1/KDR) That Inhibits Tyrosine Kinase Catalysis, Tumor Vascularization, and Growth of Multiple Tumor
Types. Cancer Res. 1999, 59, 99-106. Pubmed ID: 9892193
2. Mendel et al., The Angiogenesis Inhibitor SU5416 Has Long-lasting Effects on Vascular Endothelial Growth
Factor Receptor Phosphorylation and Function. Clin. Cancer Res. 2000, 6, 4848-4858. Pubmed ID: 11156244
3. Mezrich et al., SU5416, a VEGF Receptor Inhibitor and Ligand of the AHR, Represents a New Alternative for
Immunomodulation. PLOS One, 2012, 7(9), e44547.
|
|